
Policy Portfolio
Friends of Cancer Research Policy Portfolio
Friends of Cancer Research (Friends) fosters collaboration among leaders from federal health and regulatory agencies, academia, the private sector, and patient advocacy groups to identify and address critical issues in development of new oncology drugs. By bringing together diverse perspectives, Friends proposes unique approaches to solve current challenges in oncology drug development. The evidence and proposals generated from these efforts inform Friends’ legislative and regulatory policy priorities, which are organized into specific focus areas including:
Advancing Drug Development
- Expedited Programs
- Tolerability and Dose Optimization
- Novel Endpoints

Inclusive Clinical Research
- Eligibility Criteria
- Diversity Action Plans
- Pediatric Drug Development
Diagnostic Harmonization and Development
- Diagnostic Test Development
- Transparency in Test Performance
- Artificial Intelligence/Machine Learning in Diagnostic Tests

Innovative Trial Designs
- Real-World Data & Evidence
- Cell and Gene Therapies
- Pragmatic Trials
Advancing Drug Development
Friends works to ensure oncology clinical trials are designed to be patient-centered and employ streamlined methods to generate rapid evidence supporting safe and effective therapies.
Expedited Programs
Expedited programs at the U.S. Food and Drug Administration (FDA) facilitate timely development and review of therapies that address unmet medical needs and treat serious or life-threatening conditions. Programs such as Fast Track, Breakthrough Therapy designation (BTD), Priority Review designation, and Accelerated Approval are instrumental in enabling earlier access to innovative therapies while maintaining rigorous approval standards. Friends recommends several policy actions to support optimal use of expedited pathways.
Policy Actions and Opportunities:
FORMALIZED FRAMEWORK FOR ACCELERATED APPROVAL:
Establishing a structured framework for sponsors to engage with the FDA early in the development process to enhance the effective use of the Accelerated Approval pathway. This framework would encourage focused discussions on key elements, including eligibility for accelerated approval, the use and development of surrogate or intermediate clinical endpoints, clinical trial designs, and plans for confirmatory trials to ensure timely and efficient development and review.1,2
Read More
Currently, there is no formal planning process for product development through the Accelerated Approval pathway, which can lead to inefficiencies and a lack of clarity around the optimal approach to designing studies and using this pathway.
CROSS-AGENCY COORDINATION:
A coordinated process between federal agencies (e.g., FDA and CMS) would avoid delayed coverage for therapies leveraging expedited development programs.3
Read More
Delays between an expedited approval decision and CMS coverage can arise, especially for novel therapies, due to complexities in establishing coverage and reimbursement processes. Novel therapies leveraging an expedited development program through FDA should prompt early engagement with CMS, particularly for products that present new challenges or uncertainty about coverage.
Read More
More than half of expedited approvals use a combination of expedited programs which can lead to redundancies in administrative requirements across programs.
Resources
Tolerability and Dose Optimizations
As novel therapies are developed and potentially administered to patients for longer periods of time, ensuring tolerability and identifying the optimal dosage have emerged as critical priorities in drug development. Since 2013, Friends has convened experts across government, academia, advocacy, and industry to discuss strategies for optimizing oncology drugs dosing, emphasizing the importance of incorporating the patient voice in the assessment of tolerability. Friends recommends several policy actions to support identifying an optimized dosage in oncology drug development.
Policy Actions and Opportunities:
Read More
The continued focus on identifying and using the maximum tolerated dose (MTD) in oncology often stems from a desire for speed and assumptions that higher doses are more effective, which may increase toxicities and prevent patients from benefiting from new treatments.
CLINICAL PHARMACOLOGY SNAPSHOT:
When submitting a novel oncology therapy for review, sponsors should consider using a drug development snapshot template focused on dose and administration that provides key areas of consideration and supporting evidence for the proposed dose and schedule to facilitate the exchange of key information between sponsors and the Agency.8 Friends worked with sponsors to develop a template to inform considerations for oncology dosing. Using such a template can help facilitate exchanges around key considerations to support evidence generation for dosage optimization and helps plan content for meetings between sponsors and FDA.9
Read More
Identification of challenges with dosage optimization may not align with milestone meetings that are typically attended by cross-functional groups within the FDA and the sponsor drug development team, which can result in delays addressing these issues. Early identification of rate-limiting aspects serves as a vehicle to support information exchange and help determine when meetings outside of the normal milestone cadence would be most beneficial.
Resources
Novel Endpoints
As treatments improve, clinical trials that rely on overall survival as an endpoint often take longer to complete, which may delay the availability of new safe and effective drugs. Early endpoints that are reasonably likely to predict long-term outcomes can facilitate more efficient clinical trials, potentially accelerating development, approval, and patient access to new therapies through use of Expedited Pathways. Our early endpoints portfolio generates evidence to support the use of ctDNA and other early endpoints for regulatory decisions. Friends recommends policy actions to support the development and use of novel endpoints in regulatory decision-making.
Policy Actions and Opportunities:
FDA ENGAGEMENT:
Enhance FDA engagement on study designs aimed to validate novel endpoints, including establishing processes for collaborative discussions with FDA that include academic, industry, and patient advocacy groups.10
Read More
While FDA issued guidance documents on the potential for use of certain novel endpoints (e.g., pathologic complete response in breast cancer, ctDNA in early-stage solid tumors), they have not established a standardized process for validating early endpoints for regulatory purposes. Developing such a process could provide greater predictability, incentivize collaborative data collection efforts, and support the rigorous validation required to advance their use in regulatory decision-making.
Resources
Inclusive Clinical Research
Friends supports efforts to enhance diversity and representativeness in clinical trials through intentional trial design, proactive planning, and innovative patient-centered approaches. Our work has identified several opportunities for advancing equity in cancer research and care.
Eligibility Criteria
Eligibility criteria outline specific characteristics patients must have to qualify for a clinical trial. These criteria are critical for protecting the safety of patients and ensuring a reliable assessment of efficacy. In cancer clinical trials, certain exclusion criteria, such as poor performance status, the presence of brain metastases, and extensive prior therapies are commonly applied in cancer clinical trials. However, these criteria may not always be necessary and can be overly restrictive. Therefore, Friends worked with the American Society of Clinical Oncology (ASCO) to develop recommendations for modernizing several commonly used cancer clinical trial eligibility criteria.
Policy Actions & Opportunities:
MODERNIZED ELIGIBILITY CRITERIA
Commonly used cancer clinical trial eligibility criteria should be carefully evaluated and implemented to balance patient safety with inclusivity. Eligibility criteria should be supported by scientific rationale, and where appropriate, adapted to enable participation of patients that reflect the real-world patient populations likely to receive the drug.11,12
Read More
Overly restrictive eligibility criteria can slow accrual, impede efforts to diversify clinical trials, and limit the generalizability of trial results.
Resources
- 2021: Modernizing Clinical Trial Eligibility Criteria: Recommendations of the ASCO-Friends of Cancer Research Washout Period and Concomitant Medication Work Group
- 2021: Modernizing Clinical Trial Eligibility Criteria: Recommendations of the ASCO-Friends of Cancer Research Prior Therapies Work Group
- 2021: Modernizing Clinical Trial Eligibility Criteria: Recommendations of the ASCO-Friends of Cancer Research Performance Status Work Group
- 2021: Modernizing Clinical Trial Eligibility Criteria: Recommendations of the ASCO-Friends of Cancer Research Laboratory Reference Ranges and Testing Intervals Work Group
- 2017: Modernizing Clinical Trial Eligibility Criteria: Recommendations of the American Society of Clinical Oncology-Friends of Cancer Research Organ Dysfunction, Prior or Concurrent Malignancy, and Comorbidities Working Group
- 2017: Modernizing Clinical Trial Eligibility: Recommendations of the American Society of Clinical Oncology-Friends of Cancer Research Minimum Age Working Group
Diversity Action Plans
Sponsors are required to develop Diversity Action Plans for pivotal clinical trials intended to support regulatory decision-making for most medical products submitted to the FDA.13 These plans necessitate an equity-focused approach to planning clinical trials including establishing enrollment goals based on demographic characteristics (e.g., age, sex, race, and ethnicity) and clinical and other characteristics. Diversity Action Plans should describe approaches that will be leveraged to recruit and retain patients from underrepresented populations. Friends recommends data-driven approaches to ensure Diversity Action Plans achieve more equitable and representative trials.
Policy Actions & Opportunities:
COMPREHENSIVE DATA-DRIVEN ENROLLMENT GOALS:
FDA should encourage sponsors to set realistic and representative enrollment goals using comprehensive data sources, such as epidemiological studies and patient registries, and provide guidance on acceptable data sources and methodologies for combining data sources.14, 15
Read More
Different data sources may have missing epidemiologic and demographic data necessary to inform comprehensive enrollment goals. Combining multiple data sources may be needed to establish comprehensive enrollment goals disaggregated by race, ethnicity, sex, and age. Variable reporting approaches and definitions can pose challenges in combining data from disparate sources.
BROADENED ELIGIBILITY CRITERIA:
Sponsors should consider how eligibility criteria can impact enrollment of underrepresented groups and use fit-for-purpose criteria to maximize inclusivity and enrollment of diverse patients.
Read More
Overly restrictive eligibility criteria can disproportionately exclude certain patient groups, hindering inclusivity and slowing trial accrual.
IMPROVING ACCESSIBILITY AND TRIAL PARTICIPATION:
Innovative approaches and trial design elements, such as telemedicine and remote monitoring, should be leveraged to reduce barriers to participation and enhance diversity. Patient-centered approaches, such as bringing trials closer to the patient or compensating for costs associated with trial participation, can improve accessibility and enable patients to participate.
Read More
Trials are a significant commitment for patients and can be inaccessible to those not living near large academic medical centers where trials largely occur, which can disproportionately affect underserved populations.
Resources
Pediatric Drug Development
While significant progress has been made in cancer treatment and care, this progress does not always translate easily into new treatments for pediatric oncology patients. Challenges such as small patient populations, ethical considerations, and unique trial design requirements can hinder progress. Several opportunities exist to address these challenges and accelerate advancements in the treatment of pediatric cancers.
Policy Actions & Opportunities:
Read More
Small patient populations often make it impossible or impracticable to conduct pediatric studies for certain molecularly targeted agents that have improved outcomes in adult populations. Alternative approaches must be explored to generate sufficient data to support the safe and effective use of therapies in pediatric patients, particularly when small patient populations make traditional trials infeasible.
POSTMARKET PEDIATRIC RESEARCH:
Where appropriate and feasible, the FDA should continue to require sponsors conduct pediatric investigations following approval of drugs used in adult cancer patient populations.
Read More
A Friends analysis found that the passage of the RACE Act helped increase the number of oncology drugs for which pediatric investigations were conducted following approval for an adult indication by an existing loophole for drugs that have received orphan drug designation.18,19 Monitoring the progress and outcomes from these pediatric investigations is important to ensure meaningful benefits for pediatric patients, such as expanded labeling to pediatric age groups, are realized.
Resources
Diagnostic Harmonization and Development
Diagnostic tests play an increasingly important role in advancing precision medicine, particularly in oncology clinical development and patient care. Friends works to promote alignment across diagnostic tests by generating evidence to understand variability in test performance, ensuring that different testing methods are accurate and consistent for patients.
Diagnostic Test Development
Friends supports efforts to modernize the regulatory framework for diagnostic tests, including clarifying the FDA’s oversight to ensure patients receive reliable and accurate results to guide optimal treatment decisions.
Policy Actions and Opportunities:
SUPPORT FDA OVERSIGHT OF DIAGNOSTIC TESTS:
Support strategies to modernize oversight of diagnostic tests, including FDA’s authority to regulate laboratory developed tests (LDTs).20
Read More
Diagnostic tests, including LDTs, frequently inform clinical practice by identifying biomarkers that predict patient response to certain therapies and informing treatment decisions. Currently these tests are not subject to FDA regulation or oversight and may undergo varying levels of validation, which could lead to inconsistent or unreliable results.
Read More
Analytical and clinical test validation for companion diagnostic tests typically requires a robust dataset of clinical samples to ensure reliability and accuracy. However, in situations where the biomarker or the indication is rare, there are often limited clinical samples from the clinical trial, making it challenging to perform all necessary test validation studies.
RISK-ALIGNED DIAGNOSTIC TEST CLASSIFICATION:
Develop a regulatory framework or validation process to evaluate when and how to appropriately down classify diagnostic tests, ensuring that regulatory controls are in place to mitigate risks to patients and maintain test reliability.
Read More
The FDA announced23 an effort to down classify certain diagnostic tests, including companion diagnostics, based on the availability of special controls. This approach allows for the development of add-on tests through the 510(k) pathway that demonstrate substantial equivalence to already FDA-approved diagnostics.
Resources
Transparency in Test Performance
Our diagnostic harmonization efforts provide evidence on variability in assays assessing the same biomarker and actionable insights into reducing variability and improving harmonization.
Policy Actions and Opportunities:
ENHANCE TRANSPARENCY IN TEST PERFORMANCE:
Promote alignment across diagnostic tests by reporting key performance characteristics, so that patients receive consistent, accurate, and reliable test results to guide care decisions.
Read More
Projects such as the Tumor Mutational Burden (TMB), Homologous Recombination Deficiency (HRD), and Digital and Computational Pathology Harmonization (Digital PATH) Projects have identified areas for test alignment and provide evidence to support greater consistency across tests used for treatment decision-making.24
Read More
Multiple platforms are often developed for the same biomarker based on validation by different data sets, which contributes to challenges in understanding assay comparability. Common datasets can enable the demonstration of comparable performance characteristics across assays, enhancing confidence in test selection and supporting more reliable treatment decisions.
Resources
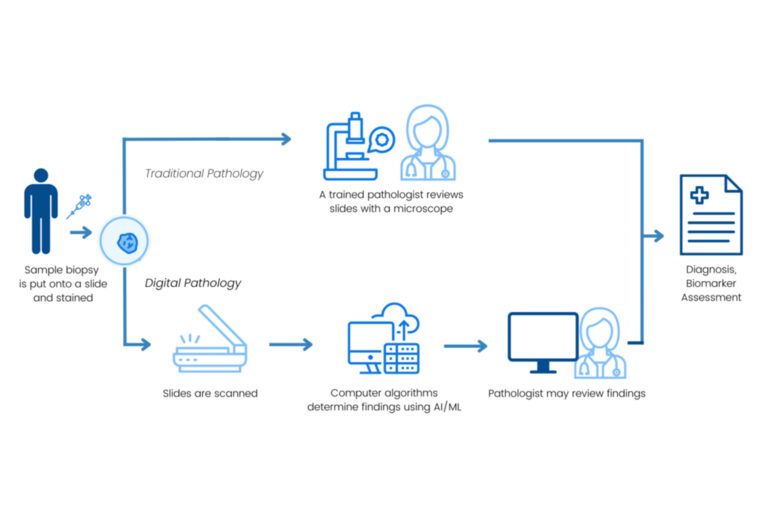
Artificial Intelligence/Machine Learning in Diagnostic Tests
Use of Artificial intelligence (AI) and machine learning (ML) in diagnostic tests has the potential to streamline drug development, enhance patient care, and improve the accuracy of diagnosis and treatment. Efforts to integrate these technologies into current regulatory frameworks are necessary to ensure the safety, efficacy, and reliability of these platforms for patients. Additionally, the adaptive nature of AI/ML technologies may require unique policy considerations that extend beyond the traditional paradigm of medical device regulation.
Policy Actions and Opportunities:
DEVELOP ROBUST REFERENCE SETS:
The FDA should establish a framework to determine the appropriateness of data sets (e.g., reliability, quality, completeness of longitudinal data, representativeness) on which AI/ML based models can be independently validated as part of approval.26
Read More
Unlike tissue and blood samples, which are often limited in supply, AI-interpretable images and related data can be banked more readily, provided proper informed consent is obtained for future use. Robust, regulatory-ready reference data sets that are representative and include necessary metadata, are lacking for the validation of models. This gap partly results from the absence of clear criteria defining a sufficient regulatory-ready reference dataset.
CLARIFY REGULATORY PATHWAYS:
Develop regulatory policies tailored to the uniqueness of AI/ML platforms. A risk-based approach driven by the intended use of the platform can help enable flexibility and efficient evidence generation to align with the benefits-risk to patients.
Read More
Existing FDA guidance does not specifically address the use of AI/ML in specific drug development contexts, such as the use of computational pathology methods in clinical trials. Regulatory expectations for the use of computational pathology platforms in oncology trials remain underdeveloped.
DEFINE NECESSARY EVIDENCE:
Develop prospective criteria and standards to guide safe development of these technologies, specifically accuracy and quality performance metrics.
Read More
Adequate evidence generation, in the form of analytical and clinical validation, is needed to support the use of AI models in oncology drug development. Performance criteria and the types of evidence needed for use of these models in clinical trials and supporting regulatory approval is unclear.
Resources
Innovative Trial Designs
Friends identifies opportunities for flexible and innovative approaches to evidence generation to streamline research, development, and regulatory decision-making, enabling broader and earlier patient access to innovative therapies.
Real-World Data & Evidence
Real-world data (RWD) collected through routine clinical care provides valuable information on safety and effectiveness in real-world care environments and can be generated more rapidly and in larger populations than clinical trial data. These data have the potential to inform regulatory decisions; however, frameworks for collecting and assessing these data must be identified to support consistent and reliable use.
Policy Actions & Opportunities:
ESTABLISH ALIGNED FRAMEWORKS FOR ASSESSING RWD:
An aligned methodology for collecting and assessing data generated through routine care is necessary to generate reliable real-world evidence (RWE). Friends has performed a series of aggregate analyses focused on establishing endpoints for oncology RWD collection,27, 28, 29 however, the field needs to align on a consistent approach.
Read More
Unlike in traditional clinical trial settings where data are collected at defined timepoints and reported uniformly for trial participants, RWD collection methods and reporting standards often vary within and across sources. Inconsistent definitions and missing data in RWD present challenges for evaluating treatment effectiveness and generating reliable insights.
DEFINE REGULATORY USES:
Increased communication and transparency around successful use of RWD in regulatory submissions can enhance existing guidance and support optimal use of RWD for regulatory decision-making.
Read More
The appropriateness of RWD for regulatory decision-making varies depending on factors such as existing data, the regulatory questions, how RWD is intended to be used. Further information and specificity around acceptable use cases for RWD would clarify FDA expectations and enable more consistent application of RWD in submissions.
Resources
Cell & Gene Therapies
Cellular and gene therapies represent a breakthrough advancement in the treatment of cancer; however, they are highly individualized and complex, requiring novel scientific and regulatory approaches for development and manufacturing. Refining existing evidence generation strategies and regulatory frameworks can enable efficient development, manufacturing, approval, and broader patient access to these transformative therapies.
Policy Actions & Opportunities:
RISK-BASED FRAMEWORK FOR DATA EXTRAPOLATION:
A risk-based framework for considering the totality of evidence from related products should be implemented to streamline development and regulatory decision-making for next generation products and facilitate earlier patient access to enhanced therapies.30,31
Read More
Cell-based gene therapy development is complex and resource-intensive. Often, sponsors develop and test multiple versions of a product in tandem or parallel, generating evidence for each version. Duplicative data generation can lead to inefficient resource utilization and slow the development of new therapies, delaying approval and patient access.
BROADENING ACCESS TO THERAPIES:
Novel approaches to manufacturing, storing, and administering cell and gene therapies are needed to support broader patient access.
Read More
Manufacturing, transporting, storing, and administering cellular and gene therapies pose several unique logistical challenges. Currently, there are relatively few sites where these highly individualized and specialized therapies are manufactured, creating barriers to patient access.
Resources
Pragmatic & Decentralized Trials
Traditional clinical trials are resource intensive for trial sites and can pose access challenges for certain patient populations. Incorporating pragmatic and decentralized trial elements such as telemedicine, streamlined data collection, and use of community oncology sites for routine trial testing, can reduce the burden of trial conduct and participation. These approaches also enhance trial efficiency and generate insights that better reflect real-world care settings.
Policy Actions & Opportunities:
Read More
Increased clinical trial complexity places significant demands on trials sites and participates and can lead to inefficiencies in trial execution and data analysis. This complexity can also create challenges in generating data that reflect real-world patient populations and clinical care settings.
