
Emerging data from clinical trials evaluating the use of immune checkpoint inhibitors (ICI) as part of adjuvant, neoadjuvant, and perioperative regimens indicate improved outcomes for patients with resectable non-small cell lung cancer (NSCLC), including early data suggesting a survival benefit. Perioperative treatment encompasses the entire period surrounding surgery, with neoadjuvant therapy administered before surgery and adjuvant therapy given after surgery. However, current two-arm trial designs fail to adequately assess the contribution of each phase of therapy (e.g., the contribution of the neoadjuvant therapy pre-surgery and the contribution of the adjuvant therapy post-surgery). As a result, there is a lack of clarity around whether the addition of ICI is necessary in both the neoadjuvant and adjuvant phase, which has generated concern around the risk of overtreatment and potential for lasting toxicities in a curative setting.
On July 25, 2024, the FDA convened the oncologic drugs advisory committee (ODAC), to discuss these issues and provide direction around the design of future clinical trials evaluating ICIs in combination regimens in the neoadjuvant and adjuvant setting for treatment of resectable NSCLC. The application and corresponding clinical trial data from patients with resectable NSCLC treated with the ICI Imfinzi (durvalumab), particularly those with stages IIA-IIIB disease without EGFR mutations or ALK gene rearrangements, were discussed.
ODAC members were asked to consider:
“In light of the uncertainty around the need for both phases of treatment, discuss whether an additional trial should be conducted to clarify the contribution of treatment phase for the durvalumab perioperative regimen prior to approval.”
The committee’s discussion highlighted that trial designs that would adequately answer this question require (Figure 1), and significant time and resources to complete, potentially delaying access to a treatment regimen that would confer benefit based on trial results from the 2-arm study (e.g. perioperative use of ICI + SOC vs SOC alone).
Figure 1: Add-On Trial: 3-Arm Design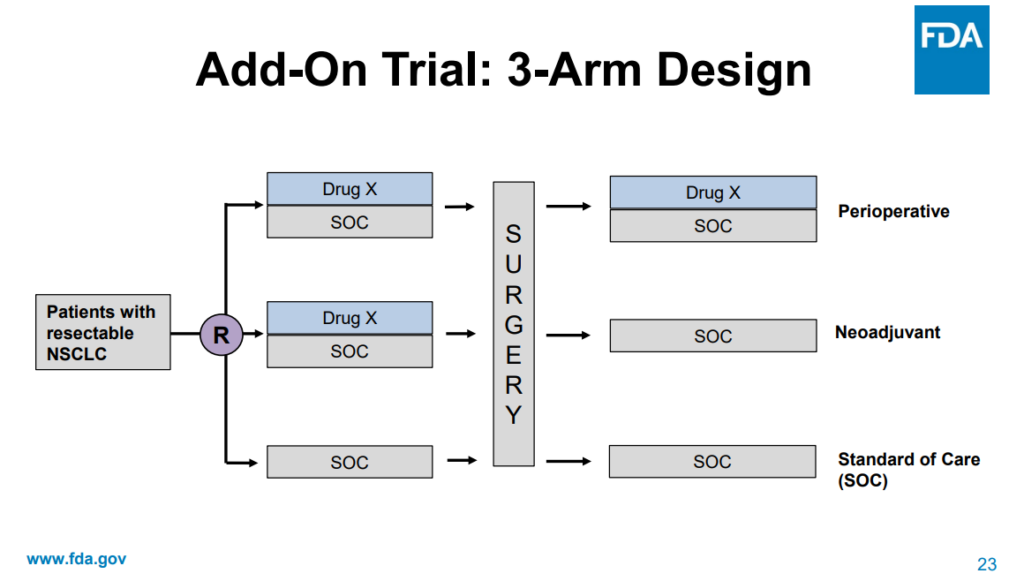 Source: Presented by FDA at the Meeting of the Oncologic Drugs Advisory Committee Meeting Announcement, July 25, 2024.
Source: Presented by FDA at the Meeting of the Oncologic Drugs Advisory Committee Meeting Announcement, July 25, 2024.
The additional trials were estimated to take between 6 and 8 years to complete. Committee members aligned with FDA that the presented 2 arm trial leaves questions about the contribution of each phase of treatment; however, they recognized that the benefit observed in the trial was sufficient evidence to support approval and provide patient access to these therapies. Following their consideration of the application, the committee unanimously voted that FDA should require that new trial design proposals for perioperative regimens for resectable NSCLC include adequate within trial assessment of contribution of treatment phase.
Throughout the meeting, ODAC members, and participants from FDA and industry reiterated the need for innovative approaches to evaluate perioperative combination regimens to balance trial efficiency with the ability to assess contribution of each phase of treatment. One approach with potential to balance these needs is a pragmatic approach to clinical trials. Pragmatic clinical trials differ from explanatory trials by incorporating design elements that reflect routine clinical practice, allowing for broader patient populations, simplified data collection, and integration into clinical workflows. During the hearing, Dr. Richard Pazdur, Director of the Oncology Center of Excellence, noted that by the time a therapy is evaluated for use in the neoadjuvant or adjuvant setting, its safety profile has been well characterized and suggested that this treatment setting provides the perfect opportunity to conduct a pragmatic clinical trial.
A second proposed approach to allow for efficient trials and timely patient access, while also ensuring trials adequately characterize contribution of phase is the use of surrogate or early endpoints, such as circulating tumor DNA (ctDNA) for assessing response to treatment. ODAC members noted there is promising data regarding the use of ctDNA for assessing response to treatment, particularly for patients with NSCLC. Early endpoints like ctDNA can potentially provide early indications of treatment effectiveness, though they require rigorous validation. Members also noted the significant work required to validate these measures, as evidenced by the ODAC meeting in March on use of minimal residual disease (MRD) as an early endpoint in multiple myeloma trials.
The ODAC hearing highlighted the continued need for innovative trial designs and novel, validated, and objective endpoints in oncology. Patient perspectives shared during the meeting underscore the importance of considering both the benefits and potential risks of new therapies, as well as the need for timely access to treatment options. Friends research partnerships, such as our ctMoniTR Project, and policy projects, such as our 2023 annual meeting working group on pragmatic clinical trials, bring stakeholders together to develop frameworks and recommendations, and generate data that advance use of these innovative drug development approaches. Ultimately, this work generates solutions that can help to resolve issues in oncology drug development, such as those discussed during this ODAC meeting, and improve outcomes for patients.
