
In 2011, Friends published a study in Health Affairs that challenged the prevailing narrative of the time, which suggested that the U.S. Food and Drug Administration (FDA) was slower and less efficient than other regulatory authorities, such as the European Medicines Agency (EMA). This study provided evidence to the contrary, demonstrating that the FDA approved novel cancer drugs more quickly, ensuring timely availability in the U.S.
Recognizing that regulatory review contributes to a small component of the time that it takes to progress a drug candidate from discovery to approval, we explored opportunities to expedite the development process for certain drugs. This work led to the establishment of the Breakthrough Therapy Designation in 2012, which identifies potentially transformative treatments and facilitates close collaboration between FDA and drug sponsors to accelerate the development and access to the most promising new medicines that address unmet needs.
In 2017, we revisited our 2011 findings, in response to reemerging criticism that FDA’s lengthy approval process was hindering the availability of treatments for U.S. patients. The 2017 analysis showed that from 2003 to 2016, 97% (71/73) of novel cancer drugs were first available in the U.S., and the FDA’s median drug review time was shorter than that of the EMA. Once again, FDA regulatory processes enabled novel cancer drugs to reach U.S. patients faster.
Now, in 2025, we have updated these data to assess whether these trends have persisted. We compared review times1 for the 152 novel oncology drugs that were approved by both the FDA and EMA between January 2003 and December 2024, excluding those withdrawn after approval. Of these 152 drugs, 143 (94%) were approved by the FDA before the EMA.
The median review time continues to be shorter for the FDA than the EMA (207 vs. 422 days), and the percentage of drugs reviewed within both six months and one year remains higher for the FDA (Figure 1). The trends reported in Friends’ original 2011 analysis and updated 2017 analysis have continued through 2024.
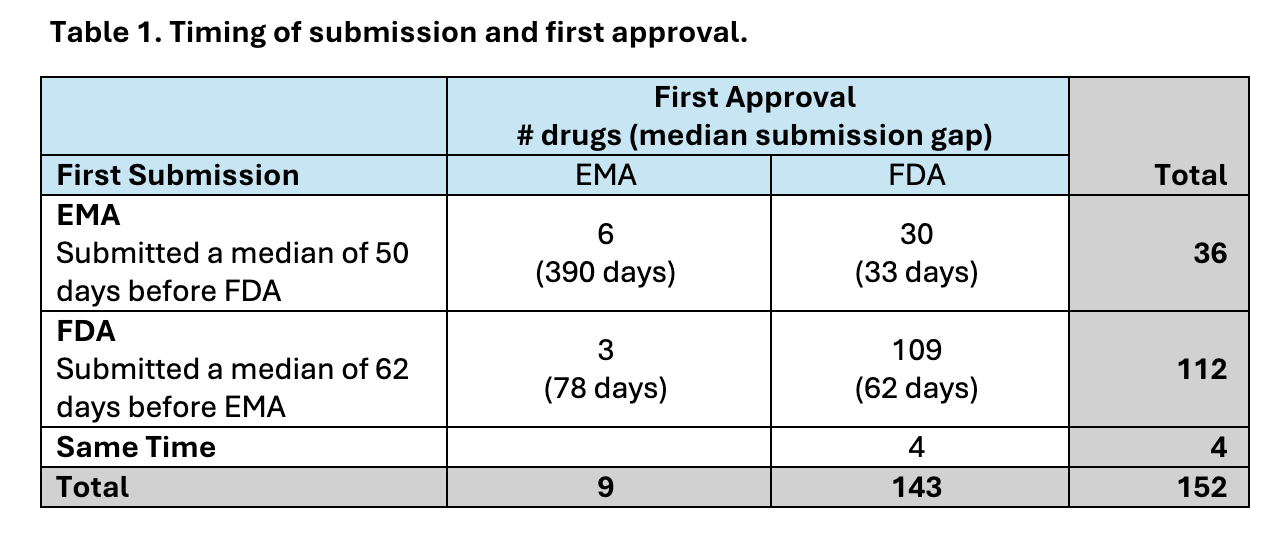
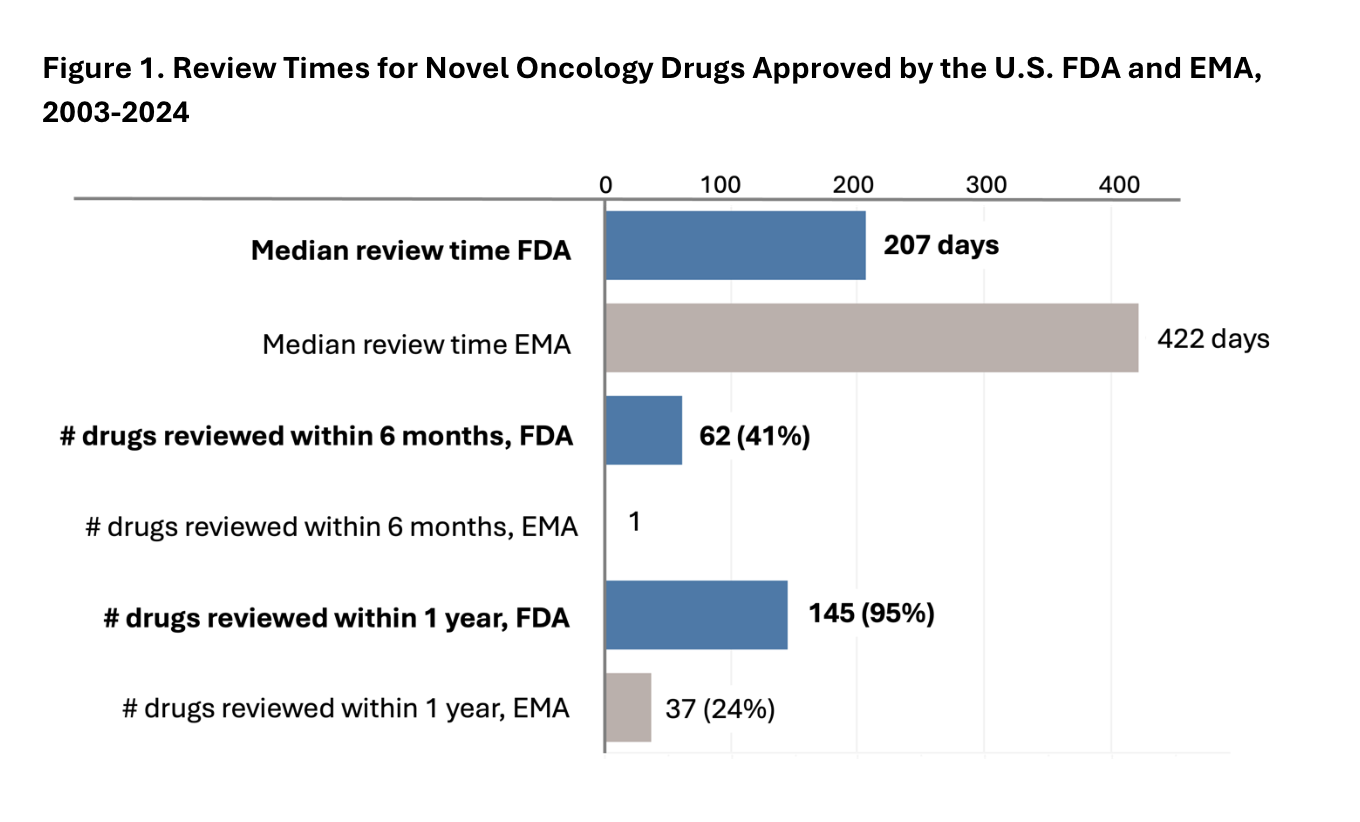 We also explored whether drug applications were submitted to the FDA before the EMA to understand whether gaps in the timing of submission contributed to earlier availability of cancer drugs in the U.S. Most drug applications (112/152) were submitted to FDA first, preceding submission to the EMA by a median of about 2 months. Even when drugs were submitted to EMA first (n=36) or submitted to both agencies simultaneously (n=4), the majority were first approved by FDA (34/40) (Table 1).
We also explored whether drug applications were submitted to the FDA before the EMA to understand whether gaps in the timing of submission contributed to earlier availability of cancer drugs in the U.S. Most drug applications (112/152) were submitted to FDA first, preceding submission to the EMA by a median of about 2 months. Even when drugs were submitted to EMA first (n=36) or submitted to both agencies simultaneously (n=4), the majority were first approved by FDA (34/40) (Table 1).
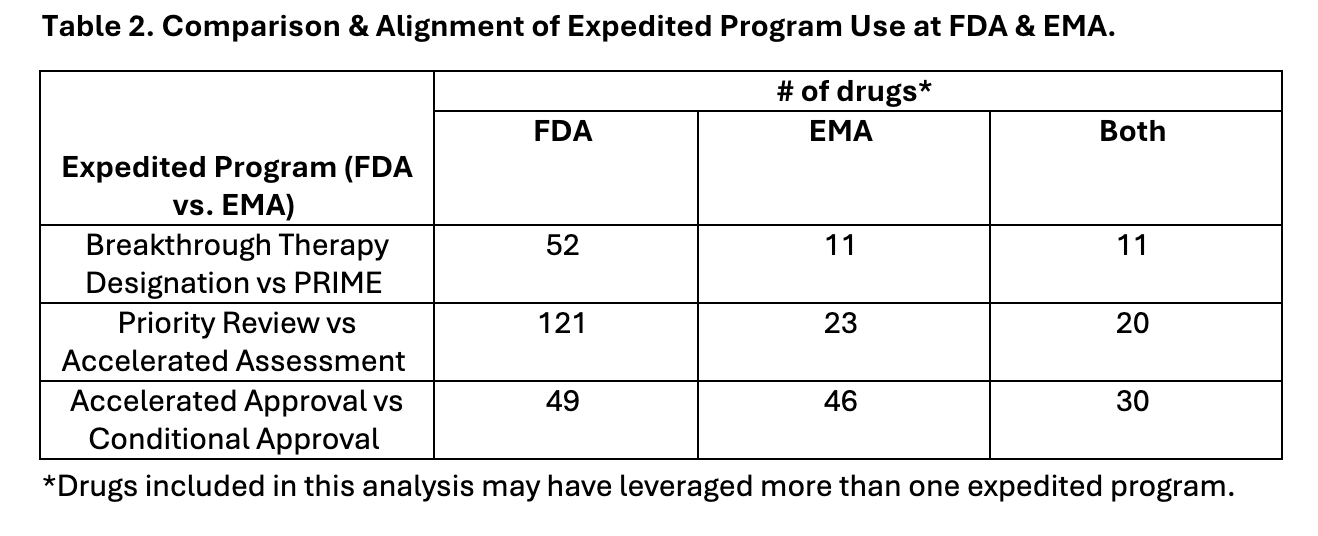
 Differences in utilization of FDA and EMA’s expedited programs could be a contributing factor to review time differences. The FDA’s Breakthrough Therapy Designation, Priority Review Designation, and Accelerated Approval programs were used more than the EMA counterpart programs (PRIME, Accelerated Assessment, and Conditional Approval, respectively) (Table 2). Both FDA and EMA programs led to more efficient reviews.
Differences in utilization of FDA and EMA’s expedited programs could be a contributing factor to review time differences. The FDA’s Breakthrough Therapy Designation, Priority Review Designation, and Accelerated Approval programs were used more than the EMA counterpart programs (PRIME, Accelerated Assessment, and Conditional Approval, respectively) (Table 2). Both FDA and EMA programs led to more efficient reviews.
Overall, these updated findings demonstrate that the FDA continues to review and approve novel oncology drugs faster than the EMA, oncology drugs are submitted for review earlier at the FDA, and drug sponsors utilize FDA’s expedited programs more frequently than the EMA. These factors, in part, likely contribute to novel oncology drugs reaching patients more quickly in the U.S.
These findings underscore the importance of continuous evaluation and optimization of FDA regulatory processes. Support for these regulatory processes will enable the FDA to continue to serve as a global leader in efficient approvals of novel drugs, particularly those addressing unmet medical need in the treatment of a serious or life-threatening conditions.
Given FDA’s leadership in establishing novel regulatory frameworks and regulatory processes, FDA can play an important role in supporting global coordination in drug review and approval processes. Efforts such as Project Orbis, launched in 2019, exemplify international collaboration by streamlining the review of oncology drugs across participating agencies. There are 13 Project Orbis Partners, including Health Canada, Australian Therapeutics Goods Administration, and Swissmedic. This initiative, which included fourteen of the drugs in this analysis, aims to harmonize and expedite global access to innovative treatments. The EMA’s recent role as an observer in Project Orbis, beginning in early 2024, highlights a growing commitment to unified global drug review practices.
Thank you to our Authors:
Bhaavna Yalavarthi is a Senior at the University of Michigan studying Public Health and Public Policy and was a undergraduate intern at Friends of Cancer Research in 2024.
Grace Collins serves as Manager, Regulatory & Data Insights at Friends of Cancer Research.
